
適應症
脊髓延髓性肌肉萎縮症(SBMA)
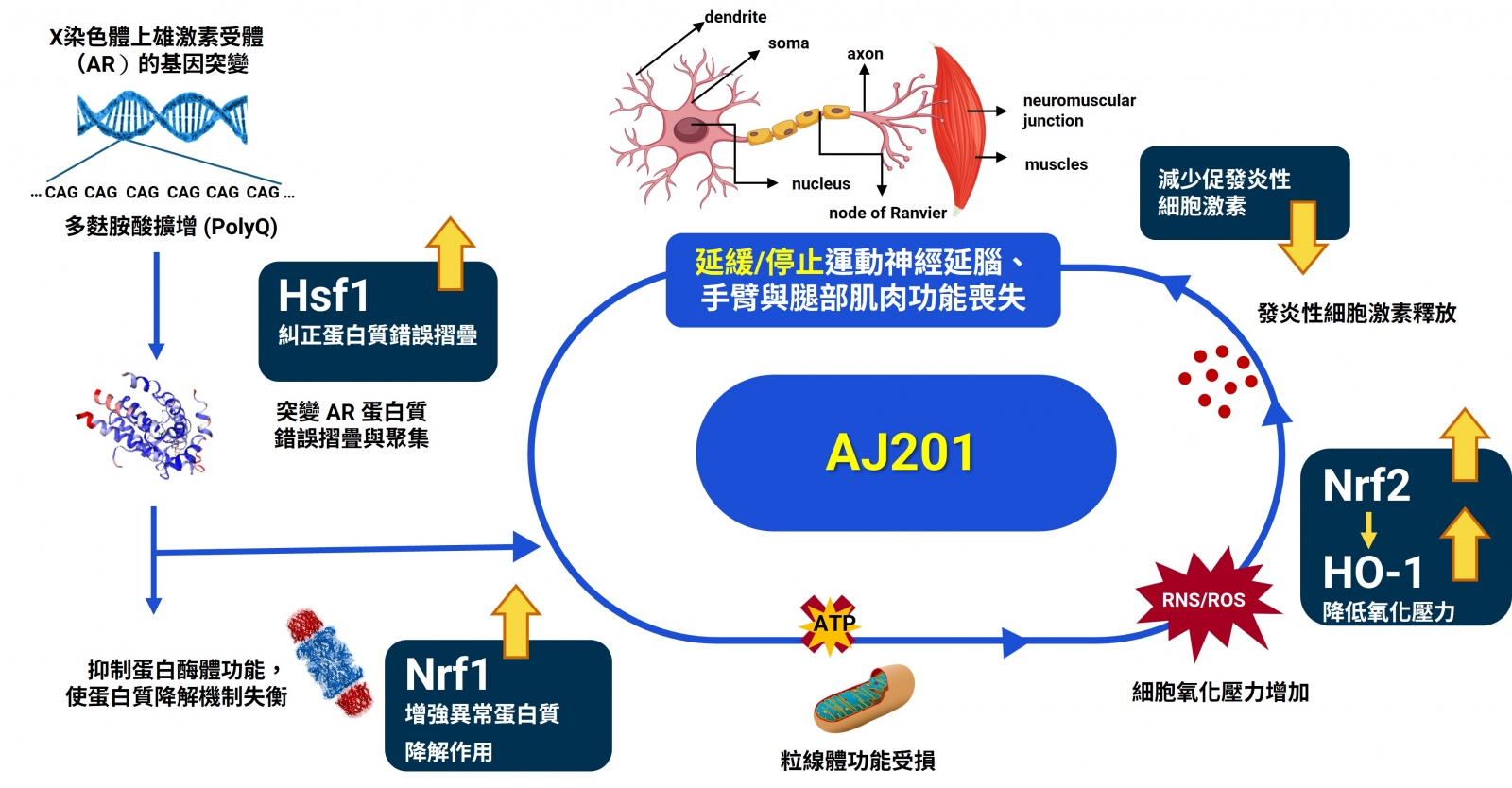
脊髓延髓性肌肉萎縮症(Spinobulbar Muscular Atrophy,SBMA),又稱甘迺迪氏症(Kennedy Disease),最早由甘迺迪醫師於1968 年提出。SBMA 是一種 X 染色體性聯隱性遺傳性疾病,其主因是雄性激素受體(Androgen Receptor,AR) 基因發生突變,第一個外顯子(exon)出現CAG 核酸片段重複數目過多(38 次以上), 形成具有過長的麩醯胺酸鍊(polyQ)的突變雄性激素受體(mutant AR, mAR)。這種mAR 堆積沉澱會引起細胞毒性、造成氧化壓力過高和慢性神經發炎,導致神經元與肌肉組織的退化和死亡。
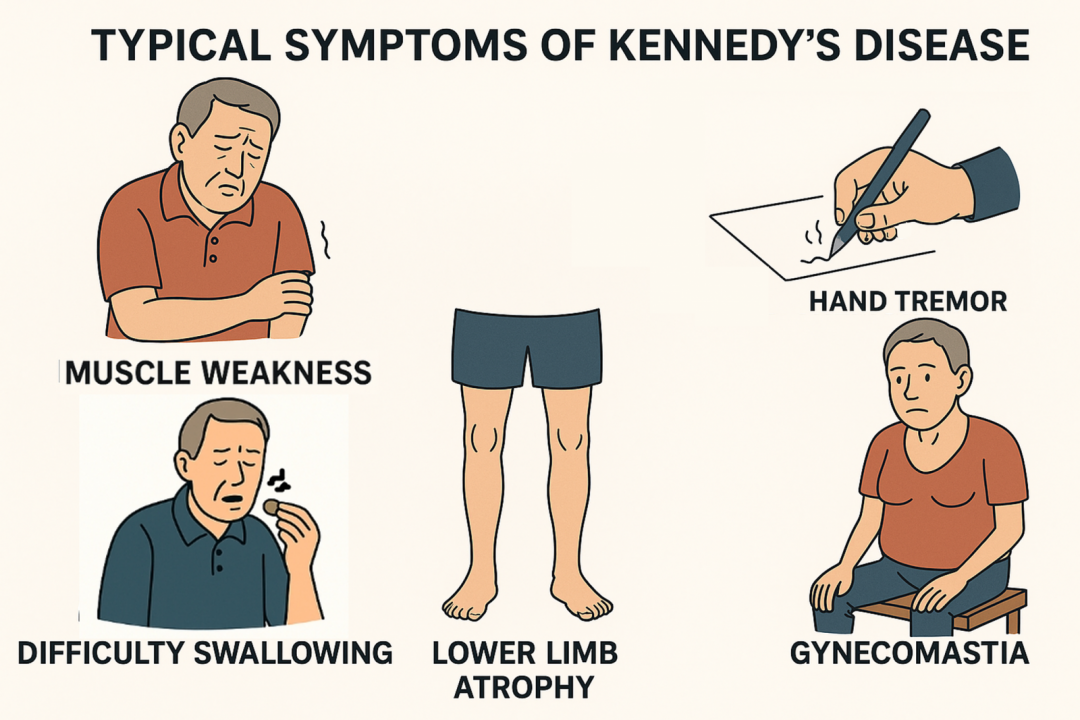
SBMA 好發於30 至50 歲間的男性,進程緩慢且具多變性,盛行率約1/40,000,為一種罕見的神經肌肉退化疾病。病人出現漸進性肌肉抽筋、無力及萎縮的現象,通常靠軀幹的大肌肉影響較為明顯,而且常以下肢無力開始,步行困難跟容易跌倒,終至失去行動能力而無法自理;喉部及舌頭因延髓部神經支配的肌肉無力導致說話含糊、咀嚼及吞嚥問題,使病人容易嗆咳,反覆吸入性肺炎是常見致命原因之一。
SBMA 好發於30 至50 歲間的男性,進程緩慢且具多變性,盛行率約1/40,000,為一種罕見的神經肌肉退化疾病。病人出現漸進性肌肉抽筋、無力及萎縮的現象,通常靠軀幹的大肌肉影響較為明顯,而且常以下肢無力開始,步行困難跟容易跌倒,終至失去行動能力而無法自理;喉部及舌頭因延髓部神經支配的肌肉無力導致說話含糊、咀嚼及吞嚥問題,使病人容易嗆咳,反覆吸入性肺炎是常見致命原因之一。
作用機制
藥理作用
AJ201可同時活化多組細胞保護路徑(包括 Nrf2、Nrf1 與 Hsf1),這些路徑與抗氧化反應、抗發炎反應及蛋白質品質控制有關,進而降低神經與肌肉損傷。
潛在市場
尚無有效治療SBMA藥物上市
孤兒藥市場的增長受到多種因素的推動,包括各國政府激勵措施,如延長市場獨占期、補助金和抵減研發費用,這些措施使孤兒藥的開發對製藥公司來說在財務上是可行的。此外,像美國食品藥品管理局(FDA)和歐洲藥品管理局(EMA)等機構的有利法規框架和加速審查流程,正在幫助孤兒藥更快地上市。同時,技術和科學的進步顯著促進了孤兒藥的開發,特別是基因組學、個人化醫療和生物技術的進步。
目前SBMA 仍無 FDA 核准之治療藥物,現行僅能透過支持性療法來改善症狀,例如物理輔具、語言治療、避免吸入性肺炎等。雖然部分臨床研究(如 Leuprorelin、Clenbuterol、IGF-1 mimetic 等)顯示在改善特定症狀 (如吞嚥功能或肌肉質量)有初步療效,但其臨床效益有限或不一致,且缺乏明確延緩疾病惡化的證據。Leuprorelin(商品名:Leuprin SR® 注射劑)於2017 年在日本獲准用於治療SBMA。該藥為促性腺激素釋放激素(GnRH)類似物,可抑制睪丸釋放睪固酮。其多項隨機雙盲臨床試驗顯示,連續48 週使用 Leuprorelin 可在部分病患中改善吞嚥功能,並減少突變AR 蛋白於陰囊皮膚的堆積。然而,其對整體運動功能改善的效果並不明確。
此外,SBMA 為罕見疾病,患者招募與研究資源有限,使得過去臨床試驗進展緩慢。儘管臨床前模型中證實多種機轉與治療標的(如雄性激素受體降解、抗氧化壓力、蛋白質品質控制),至今尚無能夠有效治療疾病或延緩疾病進程的藥物上市,顯示 SBMA 仍存在重大未被滿足的醫療需求。
專利資訊
AJ201全球專利佈局
含25 件核准公告專利以及2 件申請案,共計27 件專利/申請案分布如下圖,為安基生技100%獨自掌握,最新專利若取得專利許可,AJ201可以享有至2044年之保護期。
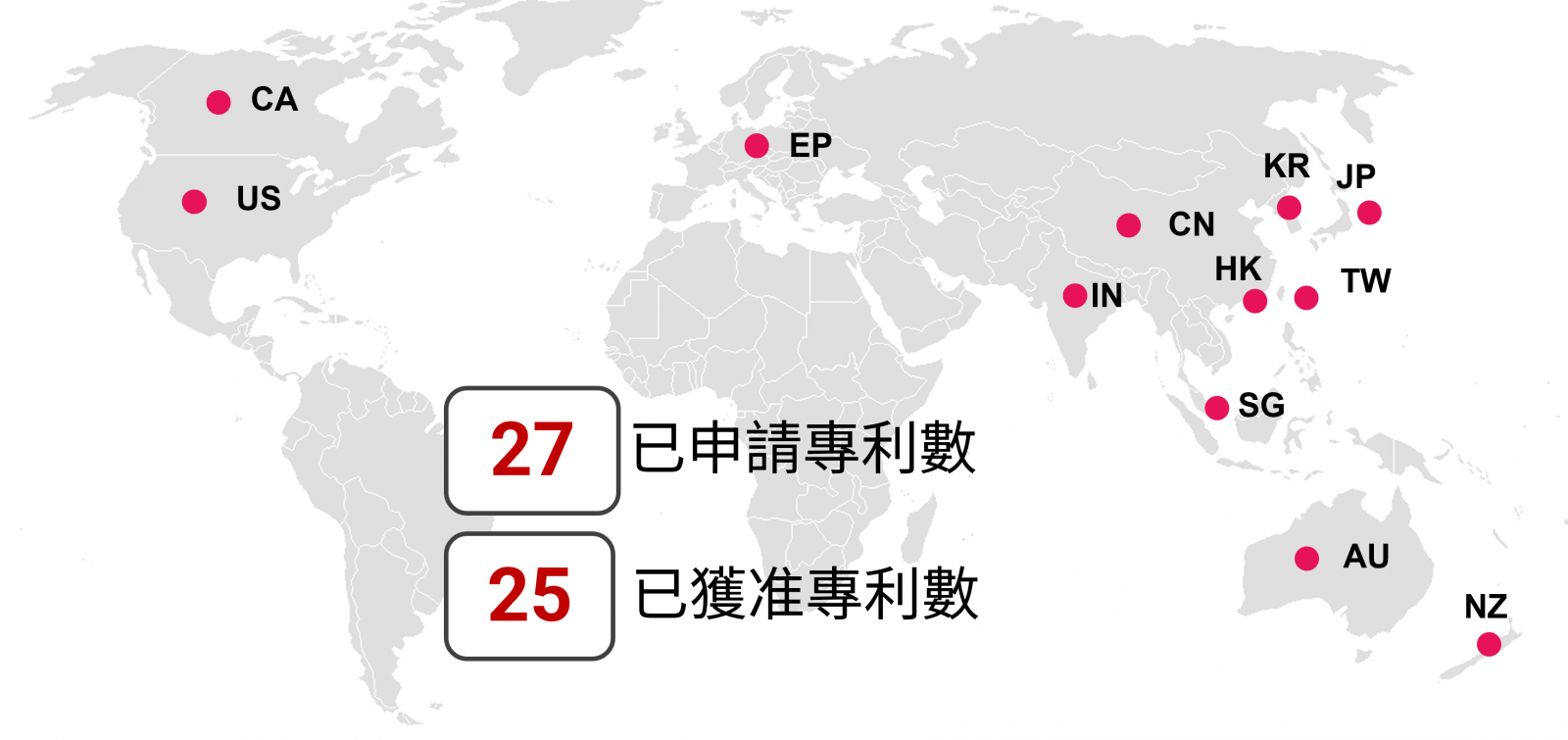
含25 件核准公告專利以及2 件申請案,共計27 件專利/申請案分布如下圖,為安基生技100%獨自掌握,最新專利若取得專利許可,AJ201可以享有至2044年之保護期。
* As of July 08, 2025